


網(wǎng)站自己上傳/AG1800.jpg)
網(wǎng)站自己上傳/AG60上傳.jpg)
網(wǎng)站自己上傳/pbs-100.jpg)
網(wǎng)站自己上傳/PBS-E.jpg)
網(wǎng)站自己上傳/IA.jpg)
網(wǎng)站自己上傳/QLC-I.jpg)
New ISO Cleanroom Standards: What Will They Mean For Pharma?
International experts have made significant changes to ISO’s airborne cleanliness classification standard—and now is the time for industry to comment.
Since 2000, industries that manufacture products in cleanrooms and other controlled environments have used ISO Standards 14644-1—Part 1: Classification of air cleanliness, and 14644-2—Part 2: Specifications for testing and monitoring to prove continued compliance with ISO 14644-1, as the global standards by which they validate the cleanliness of their cleanrooms. Last December, the ISO Working Group (WG) that developed those two landmark documents issued revised versions as Draft International Standards (DIS). Although the review and approval process for these documents to reach International Standard status will take more than a year, the DIS versions may be used now as trade references per agreement between customers and suppliers. Thus, it is imperative for those involved in cleanroom operations to understand the procedural changes contained in the Draft International Standards, as well as the reasons for those changes.
In the new ISO/DIS 14644-1—Part 1: Classification of air cleanliness by particle concentration, ISO Technical Committee (ISO/TC) 209 WG 1—the global experts who drafted the revisions—have introduced a simplified classification process that utilizes a more accurate, statistically based sampling plan. The new plan calls for a greater number of sample locations and randomized selection of those locations, allowing for different concentration levels in different parts of the cleanroom. This approach is designed to ensure with 95% confidence that at least 90% of the cleanroom area complies with the particle concentration limit.
As in the 1999 Standard, the new DIS edition of ISO 14644-1 provides nine classes of cleanliness, which specify the maximum allowed particle concentration as a function of the particle size for each class (see Table 1). Gordon Farquharson, Convenor of ISO/TC 209 WG 1, points out that WG 1 intentionally avoided making any radical changes to the principles behind the cleanliness classes. Also, as in ISO 14644- 1:1999, the new ISO/DIS 14644-1 specifies the number of sample locations for classification and the acceptance criterion for the data (see Table 2). Users will note critical differences in the content of the latter table versus the guidance for sample locations in the 1999 Standard.
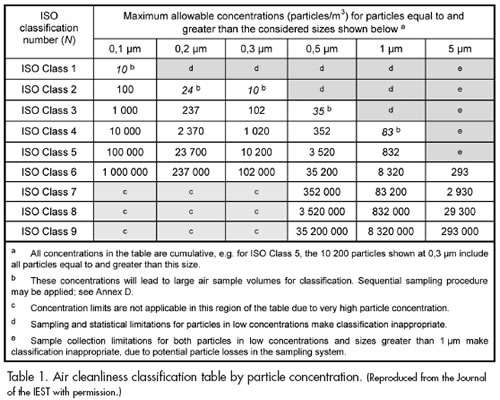
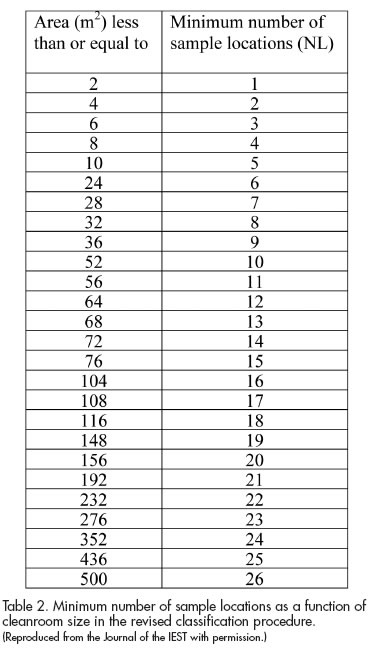
As for the companion document, the differences between ISO 14644-2:2000 and the new ISO/DIS 14644-2—Part 2: Specifications for monitoring and periodic testing to prove continued compliance with ISO 14644-1:XXXX, reflect the new sampling methodology in ISO/DIS 14644-1.
So, what impact will these Draft International Standards have on the pharmaceutical and medical device industries? The answer may change as the new documents undergo the process of becoming International Standards, but from the DIS stage forward, these documents may be specified in a business contract at any time.
The US Food and Drug Administration (FDA) has yet to weigh in publicly on the new DIS documents. However, the Division of Manufacturing and Product Quality within FDA’s Center for Drug Evaluation and Research (CDER) has released a statement saying, “FDA is supportive of efforts to set standards by organizations such as ISO, particularly when the documents are internationally harmonized. We had cited the previous version of 14644-1 in our 2004 Guidance on Sterile Drug Products Produced by Aseptic Processing. At this time we do not intend to issue a position paper on ISO/DIS 14644-1, but will review it with interest and expect to see many firms using this reference to assist in achieving CGMP compliance.”
In addition, as a member of the US Technical Advisory Group (US TAG) to ISO/TC 209, FDA will submit comments to ISO during the six-month comment period that runs through May 2, 2011. Representing a balanced input of US manufacturers, users, general interest groups and government experts, the US TAG is developing the official US position on the new DIS documents.
Anne Marie Dixon, Head of Delegation for the US TAG to ISO/TC 209, explains that developing the US position entails actively soliciting feedback from regulatory and non-regulatory stakeholders through the TAG Administrator, the Institute of Environmental Sciences and Technology (IEST,www.iest.org). “The new DIS documents represent a significant change to certification and potentially to validation,” Dixon remarks. “It is vital that anyone impacted by the ISO 14644 standards take a close look at the new drafts and the statistics behind them.”
She adds, “Standards require consensus, both in development and in practice. The comment period offers users and regulators the unique opportunity to shape these standards, which are so vital to global manufacturing and commerce.” Comments may be submitted to the US TAG via the IEST website, www.iest.org/i4a/forms/form.cfm?id=67.
WG 1 Convenor Farquharson concurs, noting that because the revised sampling plan is such a departure from the protocol in the 1999 Standard, “I suspect that ISO/DIS 14644-1 will elicit more comments than any other part of the 14644 series.”
Farquharson explains, “Some pharmaceutical manufacturers may not like the random locations aspect of the new sampling plan. Choosing your sampling locations randomly each time you measure your cleanroom’s particulate concentration introduces the possibility that you won’t get the same result this time as you did the previous time.” He adds that adopting the new approach will also require manufacturers to test more locations than before, and “they will have to tear up their current sampling protocols and rewrite them.”
Aware that the new sampling approach would likely generate some controversy, Farquharson and other members of WG 1 authored a technical paper that was published in January by IEST as a “Special ISO Edition” of the Journal of the IEST. Entitled “Sampling Plan for Cleanroom Classification with Respect to Airborne Particles,” the paper compares the sampling approaches in the 1999 Standard and the new DIS and explains why statistics dictated the change. The paper is available for download at the IEST website, and the authors are encouraging widespread dissemination to promote understanding of the revisions in the new DIS, particularly during the comment period.
Co-author of the Journal of the IEST paper and the Secretary of ISO/TC 209, Robert Mielke, suggests that drug and device manufacturers keep the big picture in mind. “Although the new DIS versions of 14644-1 and 14644-2 and the logic behind them are important, they only address part of the overall cleanroom qualification and monitoring equation for the pharmaceutical and medical device industries,” he says. “Additional tests should be performed; depending on the product that is manufactured in the cleanroom and where the facility is located, those tests may include microbiological counts, scan leak tests, air velocity measurements, and others. Regulatory agencies’ regulations and guidances, such as those issued by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), also need to be followed.” ISO/DIS 14644-1 and ISO/DIS 14644-2 are available from the IEST Publications Store at www.iest.org.
Heidi Parsons is the Technical Editor at IEST. As such, she manages Working Groups, writes articles, and edits ISO 14644 Series documents, IEST Recommended Practices, and technical papers for the Journal of the IEST.
--------------reprint from Controlled Environment
擊'開/關(guān)'")

擊這里給我發(fā)消息")

